高深度シークエンス技術とインピュテーション手法開発によるキラー細胞免疫グロブリン様受容体(KIR)多様性の解明
高深度シークエンス技術とインピュテーション手法開発によるキラー細胞免疫グロブリン様受容体(KIR)多様性の解明
Decoding the diversity of killer immunoglobulin-like receptors by deep sequencing and a high-resolution imputation method
Saori Sakaue, Kazuyoshi Hosomichi, Jun Hirata, Hirofumi Nakaoka, Keiko Yamazaki, MakotoYawata, Nobuyo Yawata, Tatsuhiko Naito, Junji Umeno, Takaaki Kawaguchi, Toshiyuki Matsui, Satoshi Motoya, Yasuo Suzuki, Hidetoshi Inoko, Atsushi Tajima, Takayuki Morisaki, Koichi Matsuda, Yoichiro Kamatani, Yukinori Okada
Cell Genomics. 2022; 2(3), 100101
坂上 沙央里
大阪大学大学院医学系研究科 遺伝統計学教室
ハーバード大学ブリガムアンドウィメンズ病院
論文のハイライト
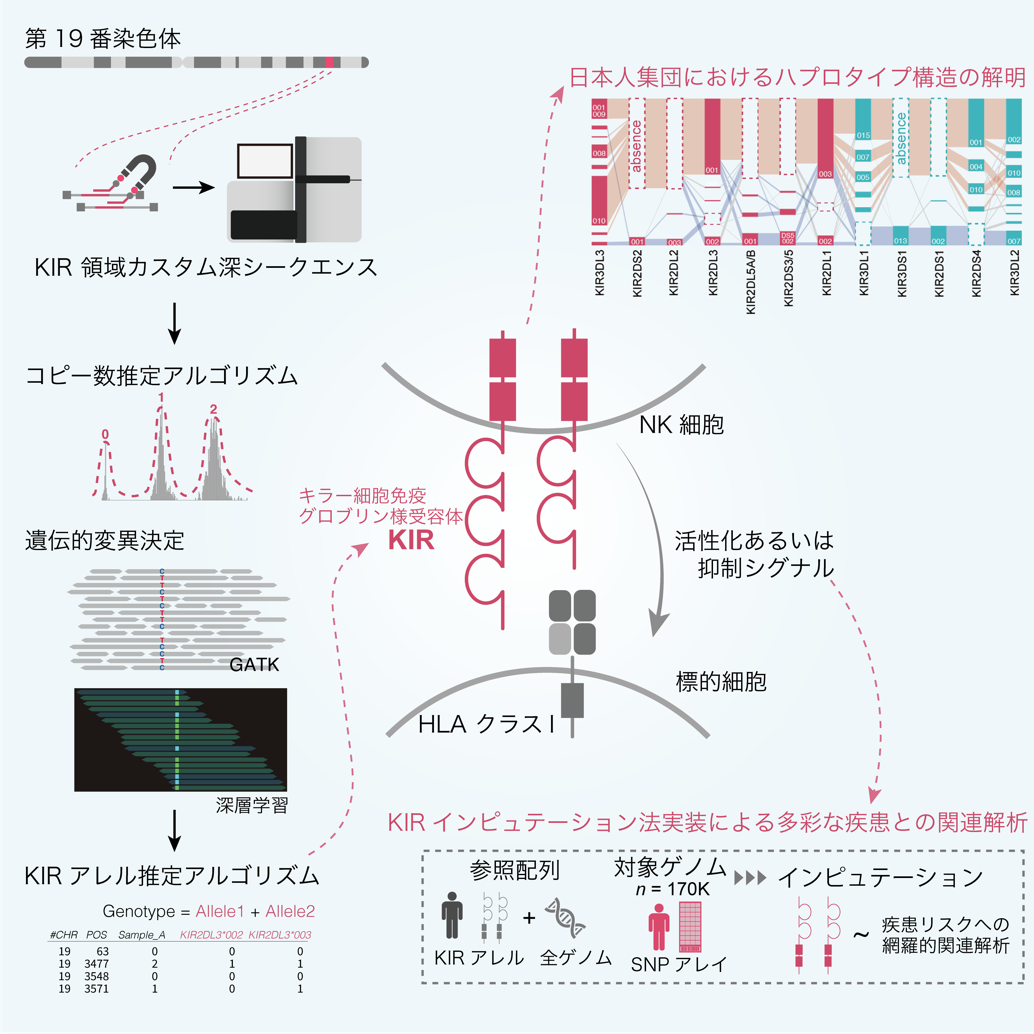
全ゲノムシークエンスが広く扱われるようになったこの数年で、ヒトの遺伝的変異と将来の疾患リスクとの関係が網羅的に明らかになった。しかし、「全ゲノム」解析と言いながらも一部のヒトゲノム領域は解析困難領域として依然謎のままだ。自然免疫機能に重要な役割を果たす19番染色体上のキラー細胞免疫グロブリン様受容体(KIR)遺伝子も困難領域の一つとして挙げられる。KIR遺伝子は、ナチュラルキラー(NK)細胞表面上に発現しHLA遺伝子を認識してその機能を調節している。そのためKIRはヒトの自然免疫応答や移植後拒絶反応に重要な役割を果たす。HLA遺伝子が多様な外来抗原や自己分子に対応するために高度な多様性を保持するように進化してきたのと同様、KIR遺伝子にも様々なレベルの多様性が保持されている。KIR遺伝子の解析が困難だったのは、領域内類似配列によるターゲットシークエンス設計の難しさや、遺伝子内の遺伝的変異数が極めて多いのみならず、個人間で遺伝子コピー数(※1)が異なり欠失挿入などの構造変異も加わり高度な多様性を構成しているからだ。しかし、造血幹細胞移植領域で移植成績にHLA遺伝子と同様にKIR遺伝子の多様性の関与が示唆されるなど、この領域の多様性を解明することの医療的な意義は大きいと考えられている。
私達は、KIR遺伝子の多様性を解析するのに特化したターゲットキャプチャー法を用いて日本人集団1,173名の高深度KIRシークエンスを実施した。さらに、カーネル密度推定(※2)による遺伝子コピー数推定、コピー数を得た上での遺伝的変異の特定、深層学習ソフトDeepVariantを用いた挿入欠失変異の特定、これらの集学的変異情報とアレル組み合わせ網羅的比較を実施し、独自のアルゴリズムを実装して高精度KIRタイピングに成功した。得られたタイピング結果を一部のサンプルで他のシークエンス手法と比較したところ、99%近い精度を得られた。
このKIRタイピング手法は非常に有用であるものの、シークエンス技術やコストを考慮すると、疾患とのKIR遺伝子型との関係を調べていくのに必要な数十〜数百万人規模に応用することは現実的でない。そこで、すでにデータが存在し安価に実施することができる一塩基多型(SNP)タイピングによるマイクロアレイゲノムデータから、KIR遺伝子型を隠れマルコフモデルを用いて推定するインピュテーション法(※3)を実装した。インピュテーションによる推定精度は99.7%と高く、これによりバイオバンク・ジャパンをはじめとするゲノムコホートの17万人を対象としたKIRアレル推定と、85の疾患・バイオマーカーとの関連解析が可能になった。KIR遺伝子領域は過去に炎症性腸疾患、乾癬や関節リウマチなどの自己免疫疾患との関連が示唆されたことがあったが、この過去最大規模の解析ではその関連の度合いは報告されていたよりも弱く統計学的に有意と判断されない水準であることが分かった。
私達はKIRシークエンス結果からKIR遺伝子型を決定する解析アルゴリズムを一般公開(https://github.com/saorisakaue/KIR_project)するとともに、日本人集団内での高精度KIR遺伝子型インピュテーション法に使用するための参照配列パネルをNBDC(バイオサイエンスデータベースセンター)にて公開(https://humandbs.biosciencedbc.jp/hum0114-v3)した。今後、感染症・免疫疾患や造血幹細胞移植の成績など、重要な医療データへの更なる応用が期待される。
※1 遺伝子コピー数
通常ヒトの細胞内の遺伝子は父親由来と母親由来の2対(2コピー)であるが、KIRをはじめとする複雑な遺伝子領域では遺伝子の重複や欠失により遺伝子が必ずしも2コピーとはならないことが知られている。たとえばKIR遺伝子領域では遺伝子により0コピーから最大4コピーまでの幅がある。そのコピー数のこと。
※2 カーネル密度推定
標本から確率密度関数を推定するノンパラメトリック手法の一つ。今回は各遺伝子のシークエンス深度と基準との比を標本点として確率密度関数を推定することで、各標本点における遺伝子コピー数の推定を実施した。
※3 インピュテーション法
KIR遺伝子型など複雑な遺伝的多型を、参照配列となる周囲の一塩基多型(SNP)と連鎖不平衡関係を用い、隠れマルコフモデルなどの数理モデルを用いて確率的に推測する遺伝統計学手法のこと。
苦労した点
この研究を始めたときには私はシークエンスデータの扱い方も知らず、そのうえKIR遺伝子内の遺伝的変異とアレルとの関係の基準データすらも整理されておらず、かなり手探りの研究でした。何度も諦めそうになりましたが、共同研究者の細道一善先生をはじめとする先生方がこれまでの研究知見やデータを快くシェアしてくださり、この複雑な領域を一歩ずつ解き明かすことができました。参照データが多く存在するわけではないだけに自分の解析手法が正しいのかどうかを自分自身で自信を持てるまでに時間を要しましたが、最終的には自信が持てるプログラムを作ることができたように感じています。今まで私が携わったプロジェクトで一番時間がかかりましたが、なんとかゴールラインまでたどり着くことができてよかったです。また、HLA/KIRタイピングについてこれまで先駆的な仕事をされていらっしゃり、今回のシークエンス結果のバリデーションも迅速に実施してくださった猪子英俊先生にも特にお礼を申し上げたいです。
研究室紹介
大阪大学大学院医学系研究科遺伝統計学教室では、幅広く大規模なhuman genetics, genomicsデータを扱い、ヒトの形質(個人差)や疾患リスクがどのような遺伝的変異によって説明され、それを用いるとどのように医療に役に立つのか、研究を行っています。私は研究室の設立初年度に参加することとなり、何もないところからDIYをしながら研究室が育っていく過程を幸運にも経験することができました。興味が発散してアクセルを踏みしめがちな私ですが、一度もそれにブレーキをかけることなく見守ってくださったPIの岡田随象先生にはとても感謝しています。

写真: 竹富島にて、左から4番目が筆者、右から3番目がPIの岡田随象先生