ロングリードを用いた局所アセンブリによるヒトゲノム反復配列の一塩基解像度でのゲノムワイド解析
ロングリードを用いた局所アセンブリによるヒトゲノム反復配列の一塩基解像度でのゲノムワイド解析
池本 滉
東京大学医学系研究科 人類遺伝学分野
Localized assembly for long reads enables genome-wide analysis of repetitive regions at single-base resolution in human genomes.
Ko Ikemoto, Hinano Fujimoto, Akihiro Fujimoto
Hum Genomics. 2023;17:21.
論文のハイライト
塩基配列決定(シークエンス)技術や多様体(挿入、欠失、逆位など)検出法の発展は著しい。全ゲノム解析時代のシークエンス技術といえば、Illuminaなどの短鎖シークエンスか、NanoporeやPacBioなどの長鎖シークエンスである。短鎖データはシークエンスエラー率が低いものの、リードが短く反復配列の解析は原理上困難である。対照的に、長鎖データは長いリードが反復配列の解析を容易にする一方でエラー率が高く精密な配列解析に難がある。多様体検出法については、究極的にはマッピング依存的な方法か、de novoアセンブリ依存的な方法に分類される。マッピング法は低深度なデータや低頻度な構造変異に対して有効な方法論であるが、反復配列に起因する誤った位置へのアラインメントによる検出精度の低下などに課題がある。一方、アセンブリ法の利点は大きな多様体を検出できる点にあるが、ハプロタイプ代表性や反復配列に起因する誤ったアセンブリが課題である。
いずれにせよ塩基レベルでの多様体の解析は重要である。短鎖や長鎖など独立な複数のデータを組み合わせることは高精度な解析に有効であるが、実際の状況においてはサンプルやコストなどの事情から多様なデータが得られないことも多い。以上の観点から、私たちはマッピング法とアセンブリ法を組み合わせ、長鎖シークエンスデータのみから精密な多様体解析を行う方法論の構築を目指した。また、開発手法を用いて全ゲノムでの多様体検出に取り組んだ。
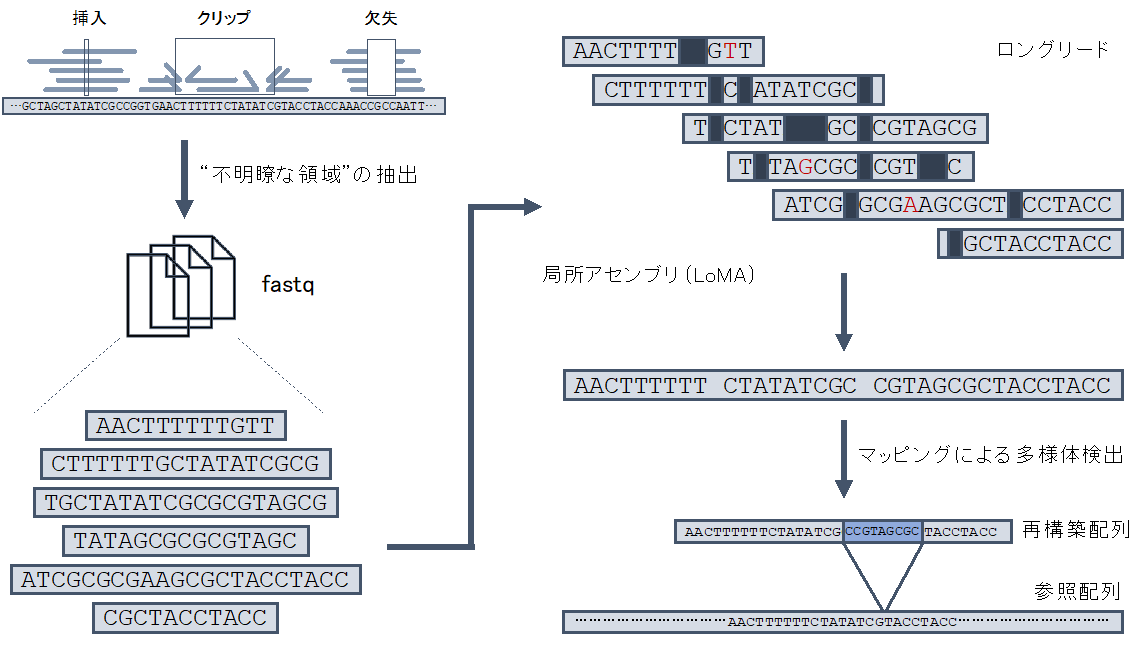
私たちは、長鎖シークエンスデータのみから高精度な塩基配列が得られるかを調べるため、まず局所ゲノムアセンブラ(LoMA)のパイプライン構築を行った。LoMAは同領域にアラインメントされたロングリードを再構築し高精度な塩基配列を出力する。アルゴリズムは、既存のminimap2とMAFFTに加え、独自のエラー除去、ヘテロ接合性構造変異の分類を組み込むことで高精度化した。Nanoporeの実データやシミュレーションデータを用いた評価では99.7%程度の高い配列精度を確認した。
次に、LoMAを用いて反復配列を中心とした領域の多様体解析を行うため、日本人(NA18943)とアフリカ人(NA19240)の全ゲノム解析を行った。初めに全ゲノムシークエンスデータ(Nanopore)の参照配列へのマッピング状態(挿入、欠失、クリップ)から“不明瞭な領域”を網羅検出した。続いて不明瞭な領域周辺にマッピングされたリードを収集した後、LoMAによって局所アセンブリすることで塩基配列を決定した。その後、藤本らが開発したCAMPHORによって再構築した配列から構造変異を検出し、特に挿入に注目した解析を行ったところ、NA18943から5516個、NA19240から6542個の挿入が検出された。多くの挿入(~80%)はタンデムリピートとトランスポゾンに由来していたが、プロセス型偽遺伝子、タンデム重複、核内ミトコンドリアDNA配列(NUMT)なども検出された。さらに、Alu要素下流やSVA内部におけるタンデムリピートの伸長など反復領域における変異の他、長い逆位、1万塩基を超える代替配列、HLA遺伝子上流における参照配列とは異なるハプロタイプといった、長鎖リード単位の解析では同定が困難な多様性が発見された。
今回の研究により、長鎖シークエンス技術を用いた、マッピングとde novoアセンブリハイブリッドな局所アセンブリ法が、ヒトゲノム複雑領域における多様体解析に有用であることが示唆された。今後は多様な領域における適用及び実証が一つの課題になると考えられる。本研究で開発した局所アセンブリツールLoMAは公開している(https://github.com/kolikem/loma)。
工夫した点、楽しかった点、苦労した点など
局所アセンブリの配列精度を担保するには、混入した不適なリードの除去や領域内反復配列をやり過ごす技が必要で、その点についてああだこうだ考える時間を持てたのは楽しかったです。振り返ると、問題設定は非常に素朴で明確でしたが、やはり難しい研究だったと思います。自分の中で納得いかない点があったせいか、とても嬉しいと予想していた初めての論文出版は、実際はとにかく出したという印象に終わりました。それでもまとめることにしたのは、研究を始めてから論文執筆の開始までに2年かかっており自分の限界を感じていたからです。そこで、今回は学問の楽しさと大変さを知るためのコンサマトリーな研究と解釈し、きっと次は学問の意義を知るインストラメンタルな研究にするのです。さて、人類遺伝学教室に入ってから藤本明洋先生には多大なる学恩を賜っております。遅くまで議論に付き合っていただいたり、研究の方向性を示していただいたり幾度となく救われました。決して綺麗な研究と言えないかもしれませんが、出版までこぎつけたのは一つの成果です。ここまで研究を導いてくださった藤本先生にこの場を借りて感謝申し上げたいです。
研究室紹介
本研究は東京大学医学系研究科の人類遺伝学教室で行いました。PIの藤本明洋先生や准教授の鵜木元香先生をはじめとするスタッフ、私を含む10名程度の学生・研究員(2023年5月現在)で構成されています(http://www.humgenet.m.u-tokyo.ac.jp)。研究室では、シークエンスデータの計算生物学的な解析によるヒト集団の遺伝的多様性・疾患(癌など)機序の解明や、エピジェネティクスが関わる生命現象や疾患の解明を目的に、日夜研究を行っています。解析系の人と実験系の人が混じっており研究テーマも多様です。私はまだ未熟者中の未熟者ですが、ユニークで面白い研究ができるように日々皆さんと一緒に頑張っています!

卒業祝いの席にて。左から2番目が筆者、左から1番目がPIの藤本明洋先生。