リンチ症候群のゲノム構造バリアント検出と切断点決定のためのナノポアターゲットシーケンス技術の適用
リンチ症候群のゲノム構造バリアント検出と切断点決定のためのナノポアターゲットシーケンス技術の適用
山口 貴世志
東京大学医科学研究所 臨床ゲノム腫瘍学分野
Application of targeted nanopore sequencing for the screening and determination of structural variants in patients with Lynch syndrome
Kiyoshi Yamaguchi, Rika Kasajima, Kiyoko Takane, Seira Hatakeyama, Eigo Shimizu, Rui Yamaguchi, Kotoe Katayama, Masami Arai, Chikashi Ishioka, Takeo Iwama, Satoshi Kaneko, Nagahide Matsubara, Yoshihiro Moriya, Tadashi Nomizu, Kokichi Sugano, Kazuo Tamura, Naohiro Tomita, Teruhiko Yoshida, Kenichi Sugihara, Yusuke Nakamura, Satoru Miyano, Seiya Imoto, Yoichi Furukawa, Tsuneo Ikenoue
J Hum Genet. 2021 Nov;66(11):1053-1060
論文のハイライト
リンチ症候群は一般大腸がんの2~4%を占める遺伝性の腫瘍で、子宮や胃などの大腸以外の臓器にも腫瘍を発生する。リンチ症候群の原因遺伝子はDNAミスマッチ修復に関わるMLH1、MSH2、MSH6およびPMS2遺伝子である。また、MSH2遺伝子の5’側上流に位置するEPCAM遺伝子の部分欠失が、MSH2のプロモーター活性の抑制を介してリンチ症候群の原因になることが報告されている。
以前、我々が行った研究で、改訂アムステルダム基準II を満たす111例のリンチ症候群が疑われる大腸がん患者についてMMR遺伝子の解析を行った。その結果62例に病的なバリアントを同定し、そのうち11例(17.7%)はMLH1またはMSH2の大きな欠失や重複などの構造バリアント(SV)であった。さらに、この11例について、ロングレンジPCRとサンガーシーケンスを用いてブレイクポイントの同定を試みたが、切断点を明らかにできたのは8例であった(Ikenoue T et al, J Hum Genet. 2019, 64(12):1187-1194)。
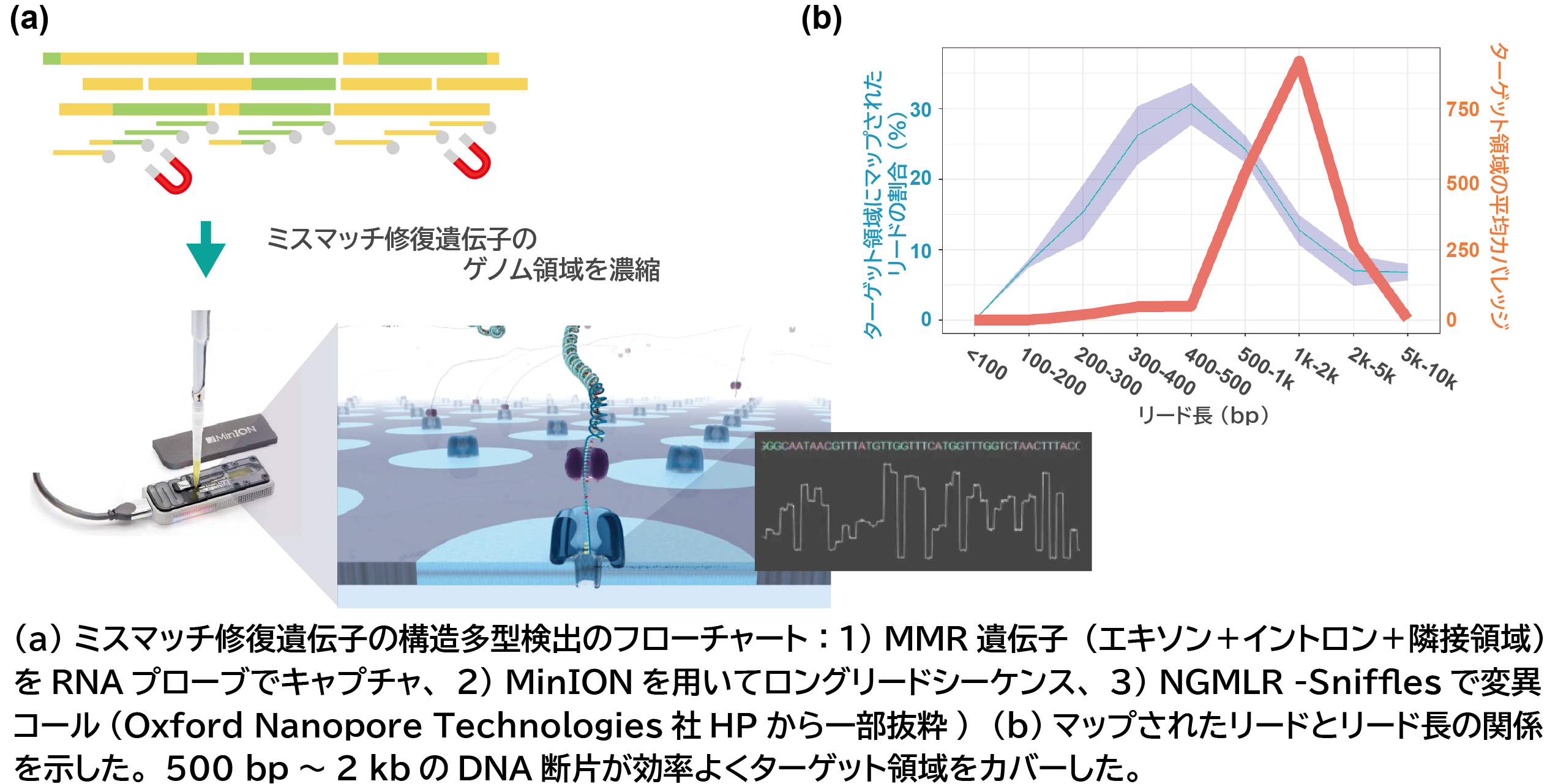
シーケンシング技術の進歩により大規模なゲノム解析が可能となり、現在、次世代シーケンサー(NGS)を用いた遺伝子パネル検査が多くの遺伝性疾患、あるいはがんの診断に用いられている。ショートリード型NGSによるパネル検査は同時に複数の遺伝子を調べることができ、主にミスセンス・ナンセンス変異(SNV)や短い挿入・欠失(short Indel)、コピー数変化(CNV)を検出できるが、イントロン内のバリアントやSVを発見することはほとんど不可能である。また、全ゲノム解析でも、大きな欠失や重複、逆位などの複雑なSVの検出は困難である。そこで、本研究では比較的安価にロングリードシーケンスが可能なオックスフォード・ナノポア社のMinIONに着目し、RNAプローブを用いたハイブリダイゼーションによるターゲットキャプチャを組み合わせて、MMR遺伝子のSV検出・切断点の同定を試みるとともに、キャプチャの際に用いるDNA断片の長さによる濃縮効率の違いについて検討した。
はじめに、MLH1に109.2 kbの欠失が同定されている症例について、MinIONによる全ゲノム解析を行った。総塩基取得数6.6 Gb、平均リード長5.6 kb、最長184.5 kbのリードを得たが、該当する切断点をまたぐ変異リードは取得できなかった。そこで我々は、MLH1、MSH2、MSH6、PMS2、およびEPCAM遺伝子のエキソンおよびイントロン、隣接領域をタイリングするように120 merのプローブを設計・合成し、ハイブリダイゼーションによって標的ゲノム領域を選択的に濃縮する戦略をとった。その結果、当該領域を平均すると約1,000倍に濃縮することができた。また、断片化DNAは2 kbまでキャプチャすることが可能で、500 bp~2 kbのサイズのDNAが効率よくターゲット領域をカバーできることが明らかとなった。そして、本症例を含む4例のMLH1またはMSH2に欠失あるいは重複が同定されている症例について解析を行った結果、すべての症例において該当するSVが検出可能であった。また、周辺の塩基配列の相同性に依存するが、ほぼ正確に切断点の位置(”Precise”: 1.0±2.0 bpおよび”Imprecise”: 142.8±131.4 bp)を同定することができた。さらに、リンチ症候群が疑われるが、MMR遺伝子のコーディングエキソン内に病的バリアントが認められない症例に対して、本法での解析を施行したところ、MSH2の一部を含む84.1 kbの大きな欠失の同定に成功した。
ナノポア技術を遺伝子診断に応用するためには、長いDNA断片を効率よく濃縮する方法の開発や、ベースコールソフトウェアの改善が必要である。しかしながら、最近では、ナノポアの一分子リアルタイムシーケンスの特徴を生かして、物理的なDNA濃縮を必要とせず、ターゲット濃縮が可能なAdaptive sampling等の新しい方法が開発されている。今後、特殊なゲノムの遺伝子解析への利用や診断技術としての応用展開が期待される。
工夫した点、楽しかった点、苦労した点など
高額な導入コストがかからないことから、気軽にMinIONによるナノポアシーケンス解析を始めましたが、導入当初は苦戦続きでした。NGS購入時のような手厚いサポートはなく、基本的にナノポアコミュニティからの情報に頼るしかありませんでした(当時)。失敗の多くは成功するまでにあきらめてしまうことが原因・・・素晴らしい名言ですが、フローセルを何枚も無駄にしました。幸いなことに、医科研内のインフォマティクス関係者のサポートもあり、その後は順調に進みました。このような恵まれた環境のなかで、新しいことに挑戦できたことに感謝しております。現在はナノポアシーケンサーを用いたDirect RNA sequencingに挑戦中です。
研究室紹介
東京大学医科学研究所・臨床ゲノム腫瘍学分野は2008年に発足しました。古川洋一教授が主宰し、現在、池上准教授と高根助教、学術支援専門職員2名、学生6名(医学系研究科・博士課程1名、新領域創成科学研究科・博士課程2名、修士課程3名)が在籍しています。大きく分けると以下の4つのテーマで研究を行っています。1)消化器がんの発生、進展メカニズム解明、2)新規発がんマウスモデルの確立と発がんメカニズム解析および治療法開発への応用、3)がんゲノム解析、4)がん分子標的治療薬の探索および開発。さらに詳しい情報は古川研ホームページをご覧ください(https://www.ims.u-tokyo.ac.jp/furukawa/)。
写真の説明:
今年(2021年)の研究室の集合写真。港区白金台キャンパスにて撮影。前列中央は古川教授、後列の右から4番目が筆者。
